Article téléchargeable au format PDF
Qu’entend-t-on par « digitalisation » des essais cliniques ?
Est-ce uniquement l’utilisation d’outils informatiques permettant de supprimer le papier ? Non, il s’agit de la dématérialisation des procédures (e-information, signature électronique, e-consent, monitoring à distance…) mais aussi du déploiement des essais en dehors des lieux classiques de déroulement (décentralisation des essais au domicile du patient par exemple, téléconsultations…).
Pourquoi en parle-t-on autant en 2023 ?
La COVID nous a forcés à revoir nos procédures et à tenter de les moderniser.
L’Europe nous pousse désormais à déployer la digitalisation.
Le contexte de digitalisation en Europe est général et dépasse la recherche en santé. Le Règlement européen Eidas appliqué depuis septembre 2018 avait déjà entrepris une harmonisation des interactions électroniques entre les citoyens et les autorités/entreprises en promouvant « le développement d’un marché de la confiance numérique » et notamment le déploiement de la signature électronique sécurisée.
La digitalisation des essais représente un enjeu de simplification et d’accélération. L’Europe s’est penchée sur cette question dans le contexte de la crise COVID afin d’assurer l’attractivité des essais cliniques dans les États, ne pas perdre de vitesse et anticiper d’éventuelles nouvelles crises sanitaires.
La France doit désormais mettre en œuvre les recommandations européennes publiées le 13 décembre 2022 (concernant tant la dématérialisation que la décentralisation des essais).
Pourquoi est-ce si compliqué ?
Comme pour tout ce qui touche à la santé, faire bouger les lignes est considéré comme risqué. Les sujets de recherche en santé sont très sensibles et renvoient aux grands principes éthiques mondiaux.
La digitalisation concerne directement les acteurs et particulièrement les personnes se prêtant à la recherche qui sont, par définition, protégées. C’est tout l’objet de la règlementation française depuis 1988 et du respect des principes éthiques nés du code de Nuremberg et de la déclaration d’Helsinki.
La digitalisation renvoie à la protection des données ultra-sensibles que sont les données de santé (et ainsi à la loi informatique et libertés de 1978 ainsi qu’au RGPD), aux questions de fracture numérique et d’égalité entre les citoyens, de protection de la cybersécurité et de confidentialité des informations concernant la santé.
Les règlementations des États européens ne sont ainsi pas toujours adaptées au déploiement de la digitalisation des essais cliniques et le travail porte désormais sur l’identification des verrous règlementaires (qui ne sont pas forcément issus de la loi mais parfois uniquement des textes règlementaires qui l’appliquent).
La France doit ainsi :
- Déterminer les modalités éthiques de l’utilisation de la e-information ou du e-consentement : prévoir un entretien face-à-face (physique ou en visio) obligatoire avant de mettre en place un e-consentement après le temps de réflexion puis un suivi totalement dématérialisé. De plus, ces procédures digitales ne sont pas intégrées dans les Méthodologies de Référence (MR) et imposent de passer par une autorisation de la CNIL supplémentaire des avis/autorisations des autorités compétentes ce qui complexifie les dossiers et allonge les délais.
- Déterminer le niveau de sécurité adéquat pour l’utilisation de la signature électronique.
- Déterminer les modalités de la responsabilité de l’investigateur principal dans la réalisation et le suivi de l’essai décentralisé : cette responsabilité implique-t-elle la présence systématique du PI dans les lieux hors du service de soin ou bien la délégation de tâches prévue dans les bonnes pratiques cliniques (décret) pourra-t-elle intégrer la notion de « sub-investigator » ?
- Cette supervision pourrait-elle faire uniquement l’objet d’un contrôle à distance ?
- Le monitoring à distance par le promoteur est-il possible et simple en France ? Il est certes possible mais sa mise en place nécessite de passer par une autorisation de la CNIL supplémentaire des avis/autorisations des autorités compétentes, ce qui complexifie les dossiers et allonge les délais (monitoring à distance non prévu dans les MR).
- Le monitoring à distance par le promoteur est-il possible et simple en France ? Il est certes possible mais sa mise en place nécessite de passer par une autorisation de la CNIL supplémentaire des avis/autorisations des autorités compétentes, ce qui complexifie les dossiers et allonge les délais (monitoring à distance non prévu dans les MR).
Où en est la France ?
Depuis février 2023 et à la suite des premières recommandations sur le e-monitoring de 2020-2021 (issues de la période d’état d’urgence sanitaire), le ministère de la santé, au travers de la Commission Nationale des Recherches portant sur la Personne Humaine (CNRIPH), mène des travaux afin d’établir des recommandations nationales précises et concrètes.
La CNRIPH coordonne ainsi un groupe de travail composé :
- de représentants des régulateurs (CNIL, CPPs, DGS, CNRIPH, ANSM, DGS, DGOS, DNS, INCa) ;
- de représentants des opérateurs (LEEM, France-Biotech, SNITEM, commission des Promoteurs institutionnels, Unicancer, ESPIC, F-CRIN, CNGE (généralistes enseignants), AFCROs) ainsi que le CNCR chargé de représenter, coordonner et porter la voix des établissements publics de santé (le CNCR a ainsi coordonné en amont et en aval un groupe de directeurs de la recherche, de pharmaciens de PUI et d’investigateurs impliqués dans la digitalisation des essais cliniques) ;
- de représentants des usagers.
Plus récemment, l’Agence de l’Innovation en Santé a rejoint le comité de coordination de ce projet.
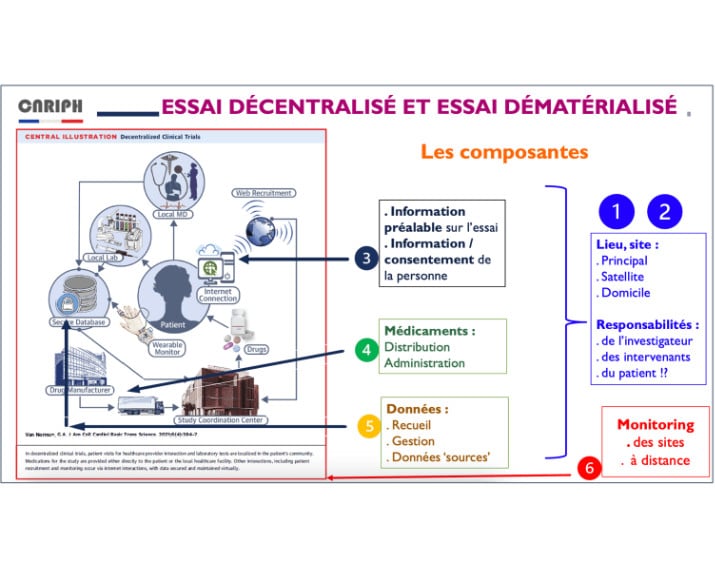
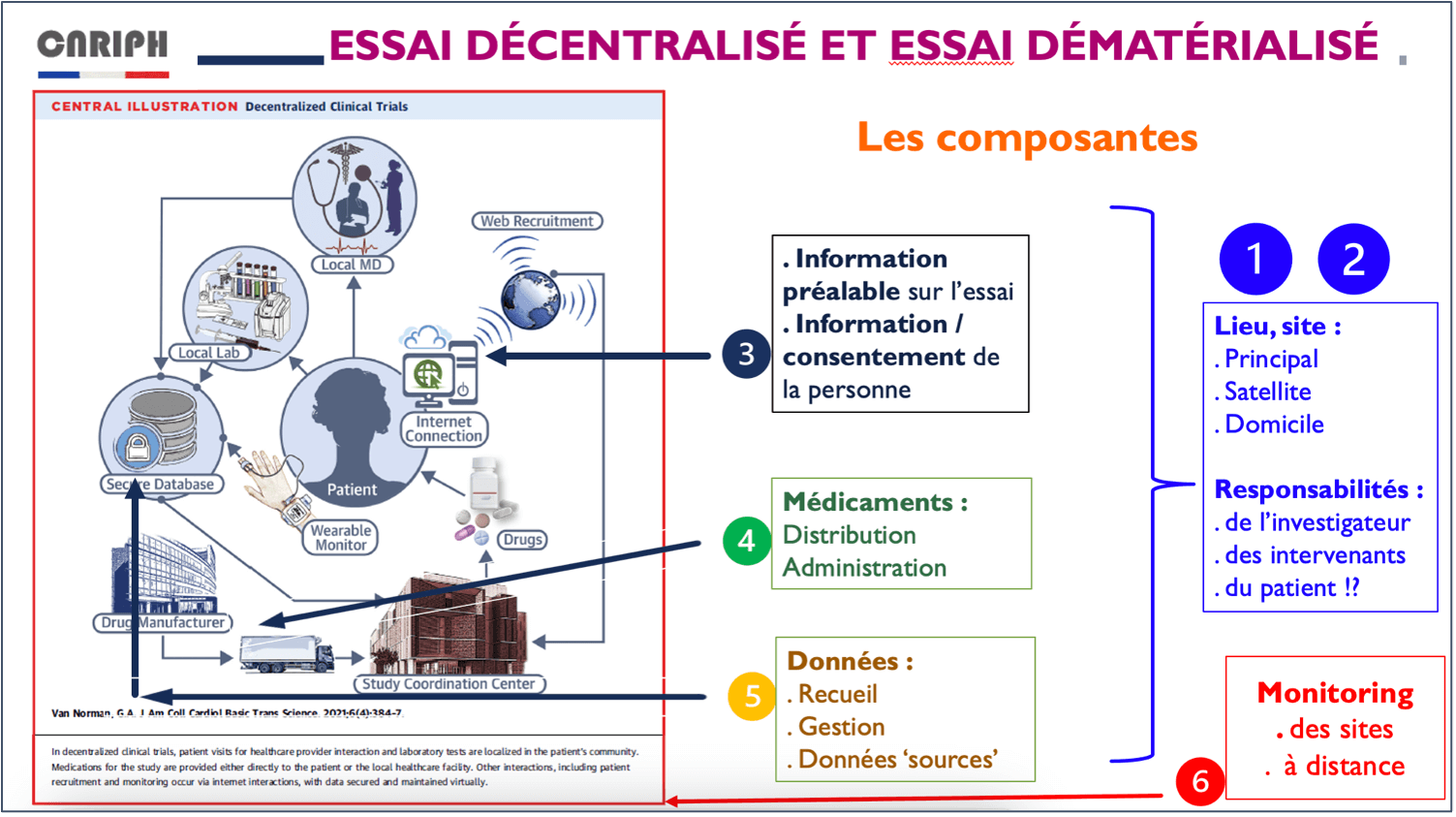
Le travail français porte sur 6 composantes des essais décentralisés :
- le lieu décentralisé ;
- la responsabilité de l’investigateur dans un lieu décentralisé ;
- l’information et consentement digital ;
- la dispensation et délivrance du médicament expérimental dans un essai décentralisé ;
- le recueil et gestion des données dans le cadre d’essais digitaux ;
- le monitoring à distance.
Une fois le travail du groupe de travail terminé, la France disposera de recommandations précises et techniques sur les méthodes et outils nécessaires à la mise en pratique des 6 composantes ainsi que des recommandations sur les verrous règlementaires à débloquer en France.
Ces recommandations seront partagées avec les États européens afin d’implémenter les recommandations européennes.
Ces recommandations ne contiendront probablement pas de propositions d’amendements à la loi. Les verrous identifiés étant règlementaires, les difficultés pourraient être réglées par la voix décrétale (modification des MR et des bonnes pratiques cliniques qui sont déjà sur le métier – non modifiées depuis 2006).
La voie de la modernisation des essais cliniques au travers de la digitalisation est par conséquent ouverte en France et dépendra par la suite de l’acceptabilité des acteurs de terrain (investigateurs, personnel de recherche clinique et patients.) Comment s’approprieront-ils les outils développés ? Ces outils seront-ils suffisamment ergonomiques aux yeux des utilisateurs ? Une chose est sûre : ils devront garantir la qualité de la relation de l’investigateur avec la personne incluse dans la recherche à l’instar de la relation médecin-patient, ADN d’une recherche en santé de qualité.


